
Recall notice
Die arterielle Kanüle wurde wegen der Gefahr einer Kabelexposition zurückgerufen, United States
vor 8 Monaten •source fda.gov
United States
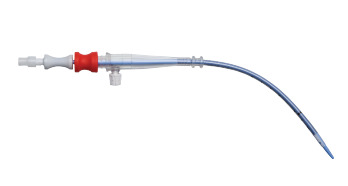
Edwards Lifesciences hat aufgrund des Risikos einer Kabelexposition einen Rückruf für bestimmte Modelle seiner arteriellen Kanüle veröffentlicht. Die betroffenen Produkte wurden landesweit in den Vereinigten Staaten vertrieben und können bei Verwendung schwerwiegende Gesundheitsrisiken darstellen. Die FDA hat für dieses Gerät einen Rückruf auf höchster Ebene herausgegeben und davor gewarnt, dass seine Verwendung zu erheblichen Verletzungen führen oder möglicherweise lebensbedrohlich sein könnte.GRUND FÜR DEN RÜCKRUF:
Edwards Lifesciences hat festgestellt, dass in einigen Fällen ein 3 mm bis 4 mm dickes Drahtsegment von der Verstärkungsspirale nahe der Spitze bestimmter OptiSite Arterial Perfusionskanülen freigelegt werden kann und sich vom Hauptkörper der Kanüle lösen kann. Da bestimmte Modelle der Oberschenkelarterienkanüle dieselben Materialien und dieselbe Konstruktion verwenden, werden diese Modelle vorsichtshalber ebenfalls in den Rückruf aufgenommen.
Die Verwendung eines Geräts mit freiliegendem Kabel kann schwerwiegende Risiken für den Patienten mit sich bringen, einschließlich erheblicher Gewebeschäden, arterieller Punktionen, die zu Blutungen führen können, unzureichender Blutfluss und Schädigung der roten Blutkörperchen (Hämolyse).
BETROFFENE PRODUKTE:
1) PRODUKT: OptiSite Arterielle Perfusionskanüle
- Modellnummer: OPTI16
- AUDI-ID: 00690103180558
2) PRODUKT: OptiSite Arterielle Perfusionskanüle
- Modellnummer: OPTI18
- AUDI-ID: 00690103180565
3) PRODUKT: Periphere femorale arterielle Kanüle
- Modellnummer: FEMII016A
- AUDI-ID: 00690103031232
4) PRODUKT: Periphere femorale arterielle Kanüle
- Modellnummer: FEMII016AS
- AUDI-ID: 00690103168341
5) PRODUKT: Periphere femorale arterielle Kanüle
- Modellnummer: FEMII018A
- AUDI-ID: 00690103031256
6) PRODUKT: Periphere femorale arterielle Kanüle
- Modellnummer: FEMII018AS
- AUDI-ID: 00690103168358
Das Problem wurde entdeckt, als Drahtabschnitte an der Kanülenspitze freigelegt wurden. Der Rückruf, der von der FDA als Klasse I eingestuft wurde, wurde am 16. Mai 2025 eingeleitet.
Wenn Sie oder ein Angehöriger verletzt sind oder Symptome haben, ist es wichtig, dies zu melden. Die Berichterstattung kann dazu beitragen, Ausbrüche frühzeitig zu erkennen und zu beheben und zu verhindern, dass andere geschädigt werden, und ermöglicht eine bessere Überwachung. Wenn die Symptome anhalten, suchen Sie einen Arzt auf.
Quelle: www.fda.gov/medical-devices/medical-device-recalls/arterial-cannula-recall-edwards-lifesciences-removes-arterial-cannula-due-risk-wire-exposure
Bemerkungen
Kommentar